The scPRINT-2 Corpus
In foundation models, data is the fuel. Language models have Common Crawl. Protein models have UniProt & PDB. For single cell, it is cellxgene: 50M cells.
But training scPRINT-2 required assembling the largest single-cell RNA-seq dataset ever used for a foundation model: over 350 million cells from 16 eukaryotic organisms, spanning more than one billion years of evolution. And building it forced us to rethink some of our assumption about how to load, sample, weight, and encode biological data at scale.
What’s in the Corpus
The scPRINT-2 corpus draws from four main sources. The backbone is the Chan Zuckerberg Institute’s CellxGene census (January 2025 release), which provides roughly 500 carefully curated human and mouse datasets with rich metadata. To this we add the Tahoe-100M dataset, Arc’s scBasecount database, and the single-cell mouse atlas of development (12.5 million cells).
The result: 350+ million unique cells, 25 TB of data, approximately 400,000 distinct genes, 4,764 different labels, and around 140,000 cell groups. The 9 metadata classes we track — cell type, disease, age, tissue, assay, ethnicity, sex, cell culture, and organism — give the model a rich supervision signal beyond just expression reconstruction.
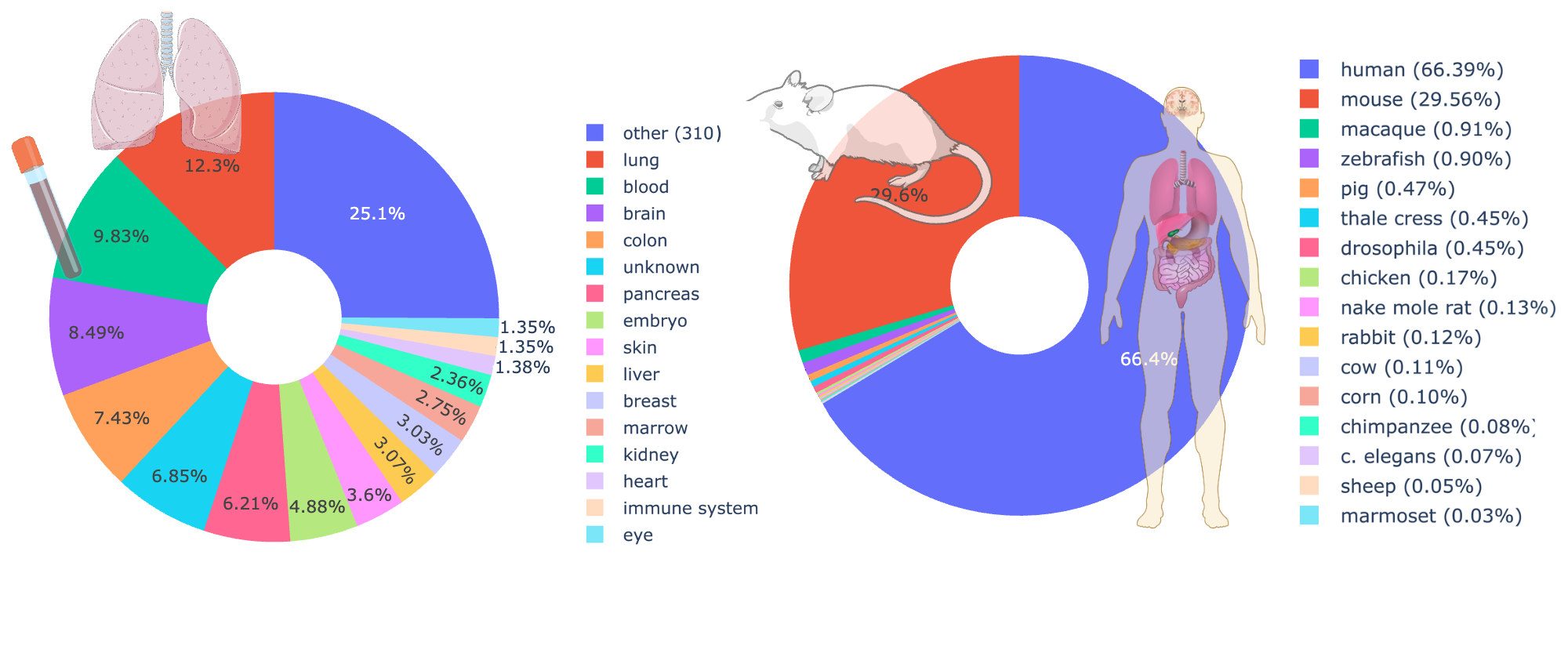
To our knowledge, this is the largest and most diverse pre-training dataset assembled for any cell foundation model. The previous generation of models (scGPT, Geneformer, scPRINT-1) were trained on 20–50 million cells, mostly human. Geneformer-v2 and STATE-SE pushed to ~160 million. We went further — but raw cell count turned out to be less important than what we found inside.
The Problem with More Data
Here is the counterintuitive result that shaped everything: adding more data does not automatically make the model better.
When we trained a model on the Tahoe-100M dataset alone — a large perturbation screen with 100 million cells — performance collapsed. The dataset has low sequencing depth and low cell-state diversity. Most cells look the same. A model trained on them learns to handle shallow, repetitive profiles and not much else.
When we added Tahoe to CxG, performance only dropped slightly for denoising. When we used all available databases, performance stayed flat. And when we reduced to just 200 random human datasets, the decrease was minimal. The conclusion is uncomfortable but clear: diversity in organisms, tissues, and cell states matters far more than raw cell count. Our current human-centric benchmarks probably undervalue this — if we could measure generalization to unseen biology more rigorously, the gap would be larger.
This also meant we had to be careful. More data brings more noise: missing annotations in scBasecount, spatial transcriptomics datasets with very low gene counts, and class imbalances that would make naive random sampling produce a model that has seen mostly common human cell types.
How We Tackled It
The core challenge was turning a raw collection of heterogeneous datasets into a training distribution the model could actually learn from. We developed three interlocking strategies.
Cluster-weighted sampling handles the datasets where cell-type annotations are missing or unreliable. Instead of relying on labels, we run Leiden clustering on the K-NN graph of each dataset and use the resulting cluster IDs as pseudo-labels for weighted sampling. This lets us apply diversity-promoting sampling even to datasets with no metadata — cells from rare clusters get sampled more often, preventing common profiles from drowning out the rest.
Depth-weighted sampling addresses the sequencing depth problem. We scale each cell’s sampling probability by a sigmoid function of its non-zero gene count, with a midpoint of 2,000 genes. Cells with more expressed genes are sampled more often — not exclusively, but enough to meaningfully shift the distribution. Before this, only 11% of sampled cells had more than 3,200 non-zero genes. After applying nnz-weighting and adding meta-cells (averaged expressions over K-nearest neighbors), more than half of input profiles exceed 3,200 genes — which is what allowed us to extend scPRINT-2’s context to 3,200 genes in the first place.
Nearest-neighbor meta-cells go beyond sampling a cell at a time. We create meta-cells by averaging the expression profiles of each cell’s K nearest neighbors (K=6). We thus sample a cell and its neighbors in the dataset by modifying our sampler to take this information into account. This gives the model access to richer, more diverse input profiles without increasing the training set size. It also encourages the model to learn from local cellular context, which is crucial for tasks like denoising and batch correction.
GNN-based multi-cell encoding goes further than simple averaging. Instead of creating meta-cells by averaging neighbor expressions, we use a DeepSet-inspired graph neural network that encodes up to 6 neighboring cells into each input token. The GNN takes raw expression values and cell-cell distances and returns a learned aggregate representation — giving the model access to local cellular context without inflating the training set size. We randomly sample between 0 and 6 neighbors per training step, so the model learns to handle variable neighborhood sizes gracefully.
Together, these methods mean that from 350 million cells, scPRINT-2 encountered roughly 2 billion distinct input profiles during training — through augmentation, variable context, and neighbor sampling. Convergence required only twice as many steps as scPRINT-1 despite a 7× larger dataset, suggesting the model was hitting diminishing returns on raw data long before we expected.
What This Enables
Training on 16 organisms — including plants, insects, fish, and mammals — gave scPRINT-2 a generalization ability that human-only models cannot match. Zero-shot, it correctly labels 42% of cells in a cat and tiger lung dataset it has never seen. It predicts mammalian organism labels for horses 72% of the time. It correctly classifies tomato plant cells into known plant organism categories despite their vanishingly small representation in training.
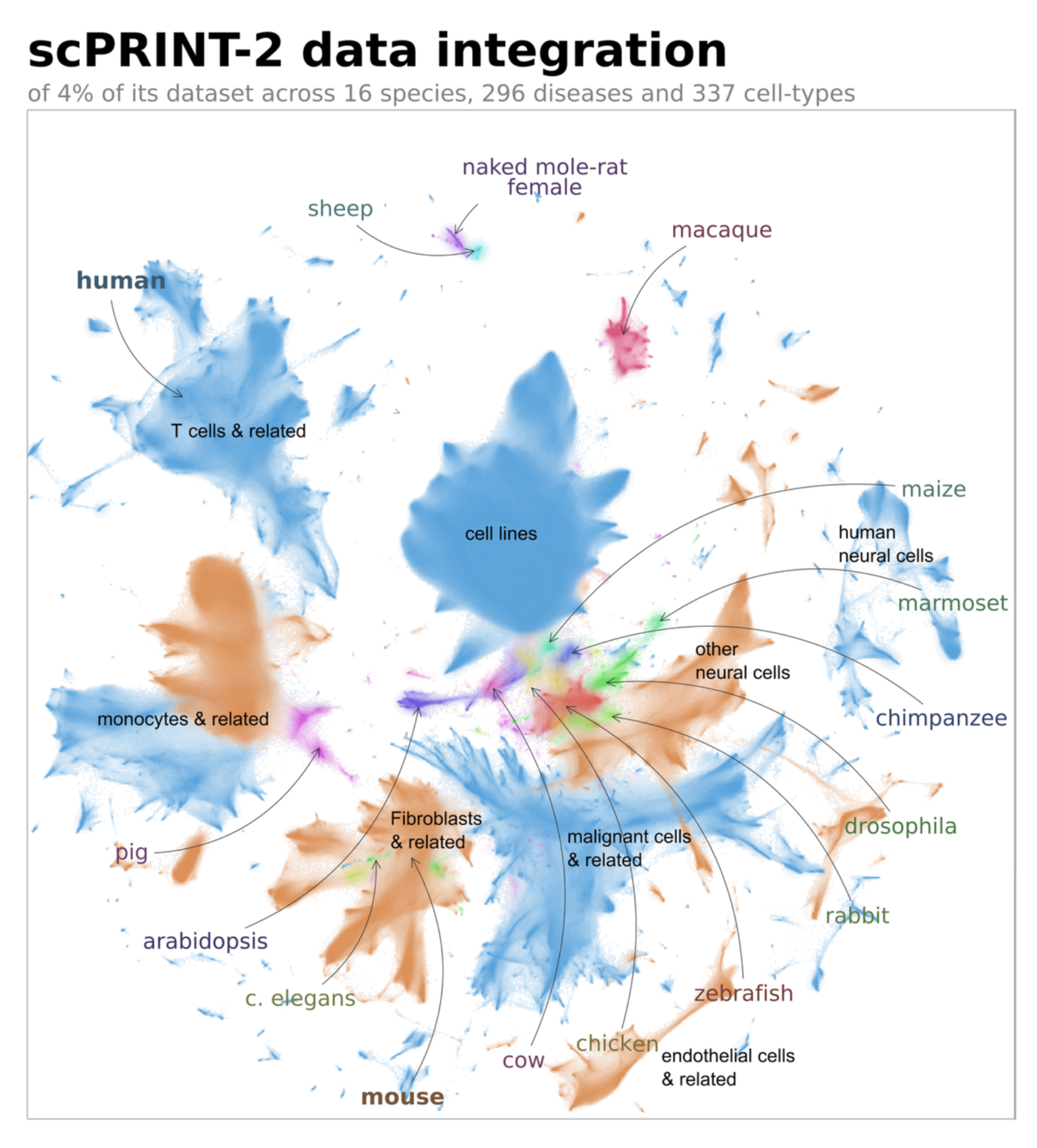
Perhaps more importantly, it can correct expert annotations. In the cat/tiger dataset, two sets of expert labels agreed only 55% of the time. scPRINT-2 flagged cells labeled as macrophages that expressed SFTPC — a known type-2 pneumocyte marker — and a differential expression analysis confirmed the model was right. The ground truth was wrong.
We have released the full 350-million-cell corpus as an aligned atlas, with scPRINT-2 cell label predictions for all classes. One percent of the atlas is available as an interactive visualization — the largest cross-organism single-cell atlas published to date.
The lesson from building this dataset is simple, even if executing on it was not: the question is never how many cells you have. It is how different they are.
scPRINT-2 is fully open-source (GPL-v3). Code, weights, pre-training data, and the full additive study (42 models) are available on GitHub. This is the third in a series of blog posts — read also about the model architecture, and Criss-Cross Attention.



Leave a comment