Introducing scPRINT-2
The field of single-cell foundation models is moving fast. In the past three years, a wave of transformer-based models — Geneformer, scGPT, scFoundation, and our own scPRINT — have been trained on tens of millions of cells and applied to everything from cell type annotation to perturbation prediction. Each paper claims state-of-the-art. Each uses its own benchmark. And almost none of them can be directly compared.
We wanted to do something different. Before building a new model, we ran a systematic abaltion study to figure out what actually works — training 42 models, each differing by a single design choice. From those results, we built scPRINT-2: a next-generation cell foundation model trained on 350 million cells from 16 organisms, with state-of-the-art performance across every task we tested.
What scPRINT-2 Is
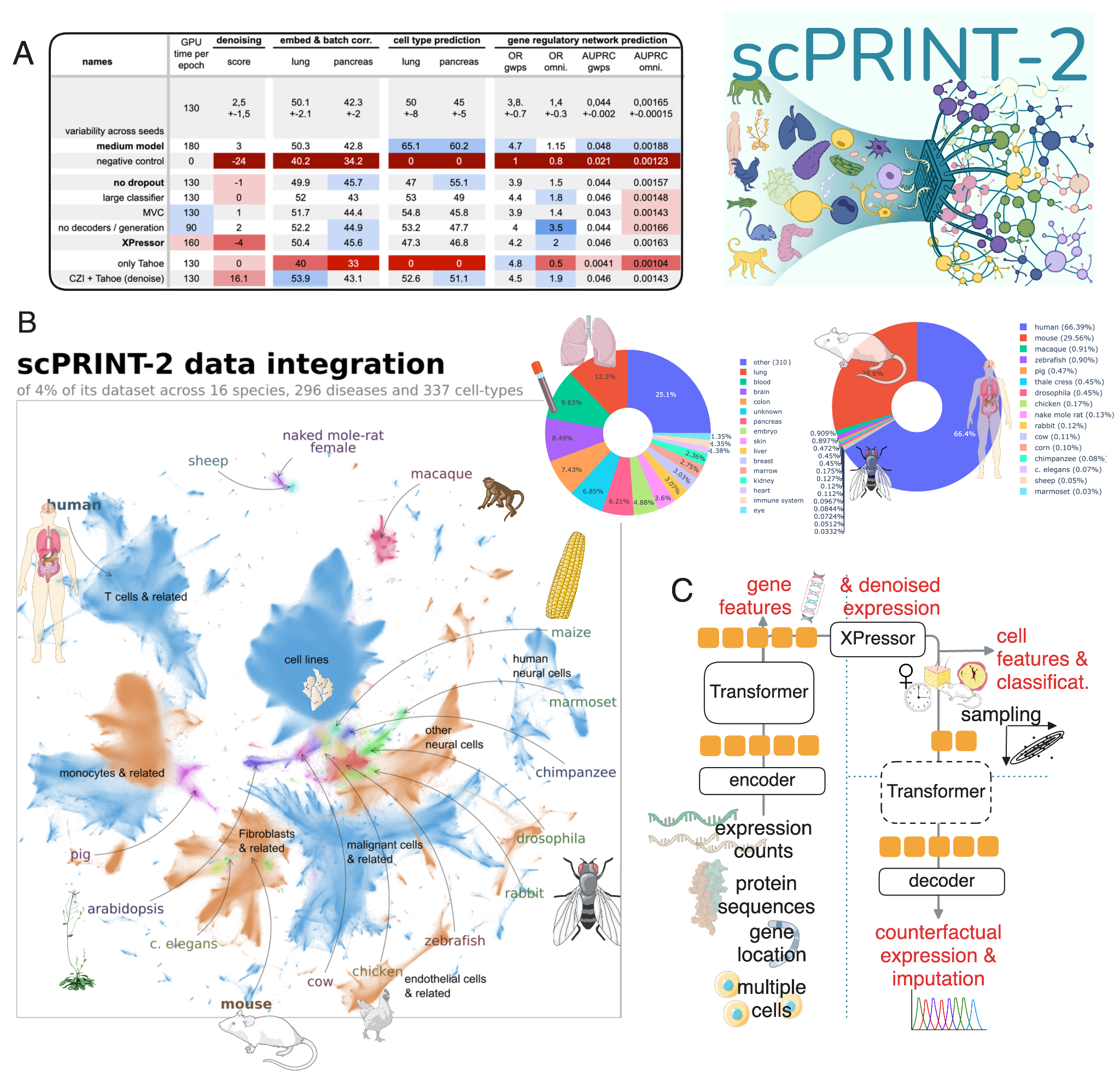
scPRINT-2 is the successor to scPRINT, which we published in Nature Communications in 2025. Where scPRINT-1 was trained on 50 million mostly human cells, scPRINT-2 scales to 350 million cells spanning 16 eukaryotic organisms, from human to mouse to tomato plant, 25 TB of unique data. It is, to our knowledge, the largest pre-training dataset assembled for any cell foundation model.
The model itself has 20 million parameters. It is semi-supervised: it leverages cell type labels and other metadata when available during pre-training, and a rich self-supervised training framework. Its outputs are rich: cell embeddings, cell type predictions across 9 metadata classes, denoised and imputed gene expression, gene-gene interaction networks, counterfactual cell generations and more.
The architecture combines a GNN-based expression encoder — which aggregates information from up to 6 neighboring cells — with gene tokens derived from a fine-tuned ESM3 protein language model representation. The transformer trunk uses bidirectional attention and XPressor compression blocks to produce compact, class-specific and regularized cell representations. Expression reconstruction uses a ZiNB+MSE combined loss. Classification is performed using a hierarchical loss that leverages the full ontological graph across 9 classes — cell type, disease, age, tissue, assay, ethnicity, sex, cell culture, and organism.
What It Can Do
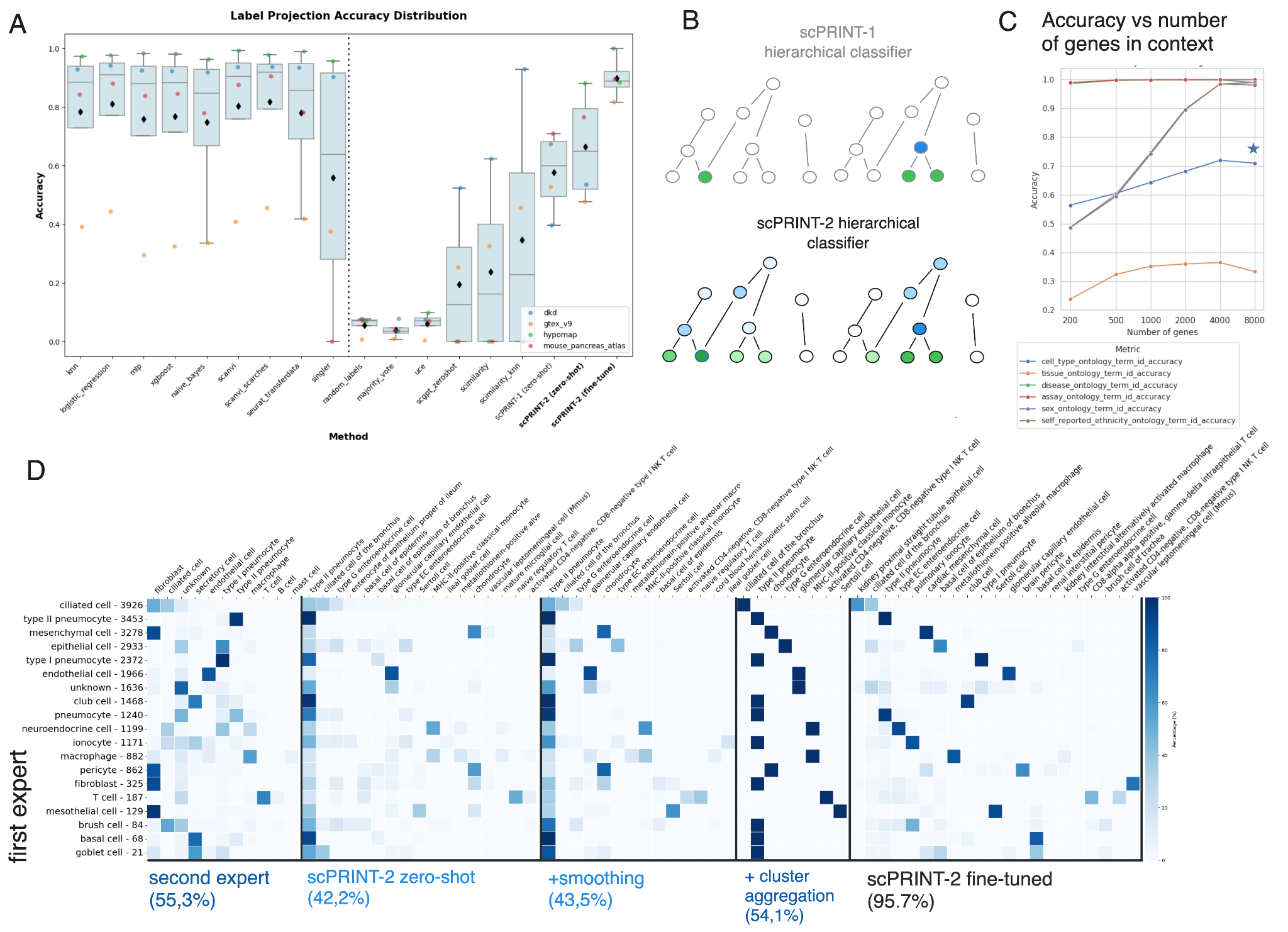
On the OpenProblems live benchmark — a competitive platform where models are tested on held-out data — scPRINT-2 achieves 75% average accuracy on cell type classification. scPRINT-1 scored 47%. Other scFMs score between 40 and 60%. scPRINT-2 is the only foundation model in the benchmark that could run on all tested datasets. Fine-tuned with our XPEFT method (parameter-efficient fine-tuning through XPressor layers only), scPRINT-2 tops every other method — supervised or unsupervised — on both classification and batch correction.
On expression denoising, scPRINT-2 is state-of-the-art across nine datasets spanning multiple sequencing technologies. It outperforms MAGIC and DCA — two methods specifically built for this task — including on the challenging regime of low-sequencing-depth cells, where scPRINT-1 struggled.
The generalization results are probably the most surprising. Using a cat and tiger lung dataset that was never seen during training, scPRINT-2 correctly classifies 42.2% of cells zero-shot — comparable to the agreement between two independent sets of expert annotations on the same dataset, which was 55.3%. Better yet, scPRINT-2 flags cells labeled as macrophages that express SFTPC, a canonical type-2 pneumocyte marker. A differential expression analysis confirms the model is right and the ground truth is wrong.
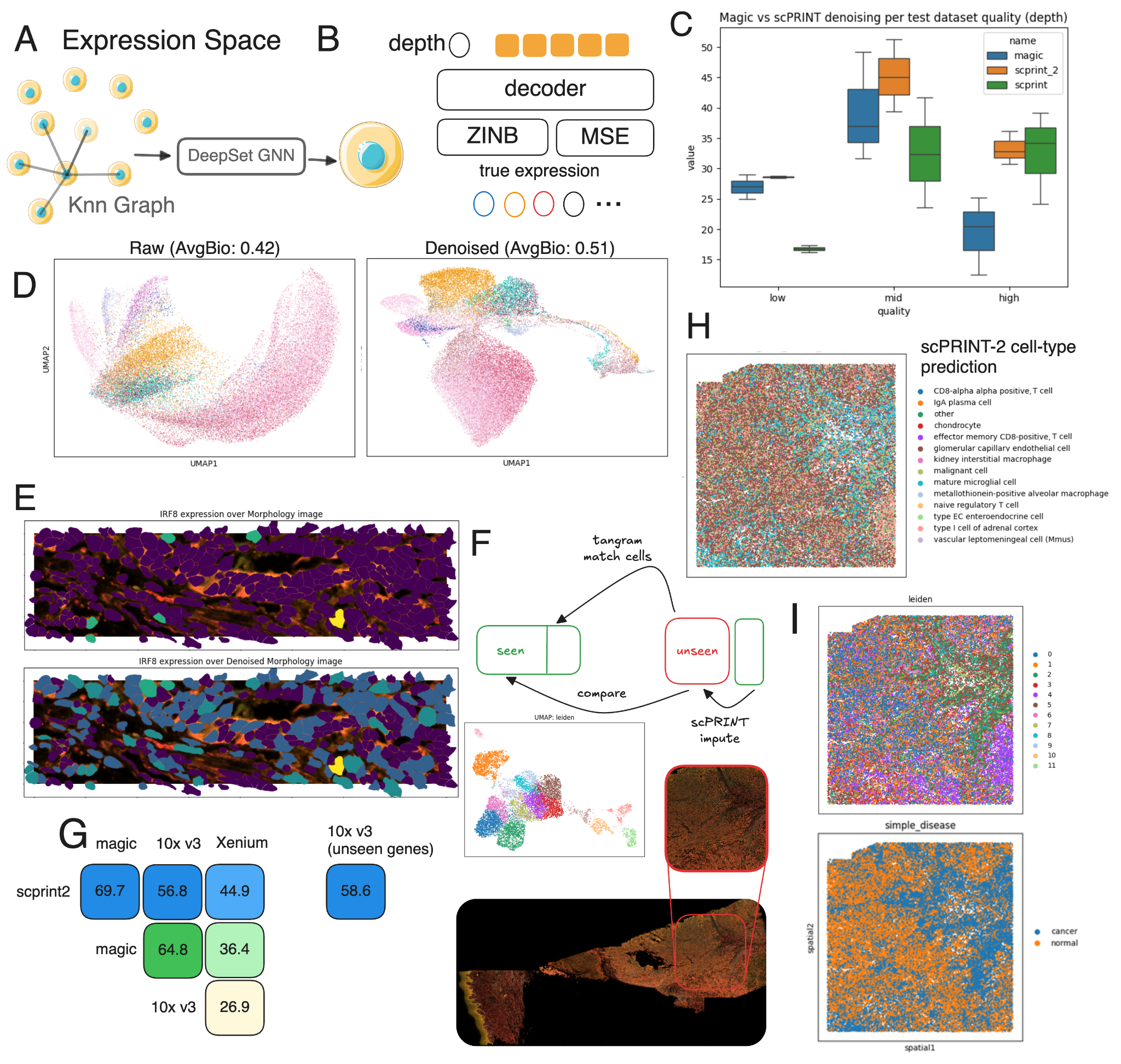
On Xenium spatial transcriptomics — a modality completely absent from training — scPRINT-2 denoises expression profiles that correlate more than 30% better with a matched 10X reference dataset than raw Xenium does. And using its generative architecture, it imputes 5,000 genes entirely absent from the Xenium panel.
scPRINT-2 can also do counterfactual generation: swap the organism embedding on mouse cells and it produces human-like profiles. 58% enrichment of correctly predicted differentially expressed genes vs random, and the pathway shifts match known biology (immune function, ECM, membrane interactions).
Gene Regulatory Networks, Zero-Shot
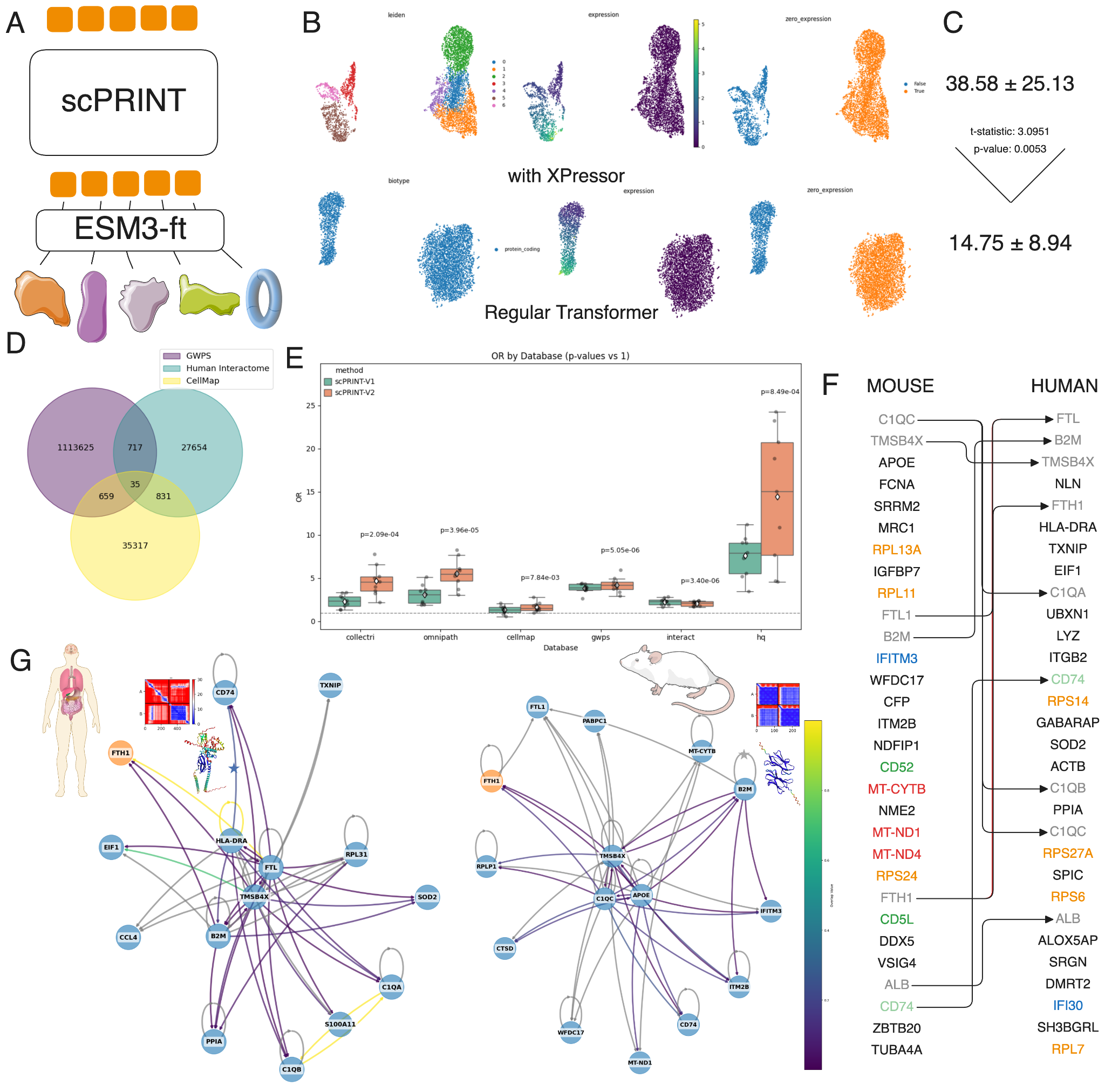
scPRINT-2 can also predict genome-wide gene regulatory networks (GRNs) directly from expression data, zero-shot. The attention patterns from pre-training encode gene-gene relationships, and we extract them as explicit networks.
This is harder to evaluate than cell type classification, because ground truth for GRNs is itself contested. Most benchmarks use regulatory databases (like Omnipath) or perturbation-based ground truth (genome-wide Perturb-seq). These disagree with each other, and both miss much of what actually happens inside a cell. We updated BenGRN to include 5 datasets across 3 metrics.
On BenGRN, scPRINT-2 is competitive with or better than existing scPRINT-1 across all evaluation regimes. But we went further and showed that you could use scPRINT-2 as a mechanism to select sets of protein-protein interactions to be assessed using AlphaFold. Pushing the idea that there might be self-reinforcement techniques to update scPRINT2 ‘s predictions using alpha fold multimer’s outputs.
What Makes It Different
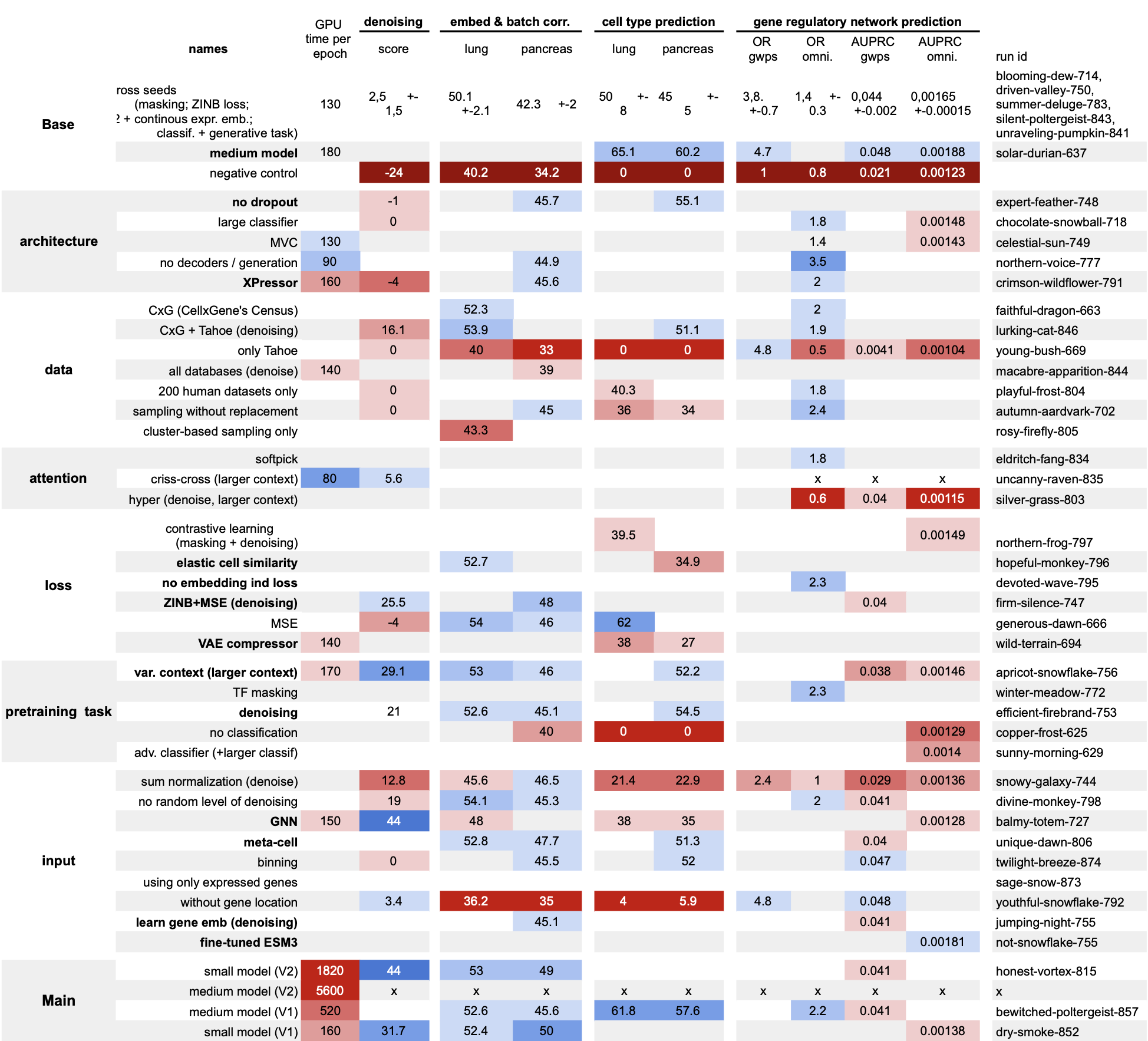
Most scFMs are trained, benchmarked, and published in isolation. Comparison across papers is confounded by dataset choices, evaluation protocols, and model sizes that all vary at once. We addressed this by running a systematic additive study first — 42 models, one changed feature each — so we know which choices actually drove scPRINT-2’s performance. The findings directly shaped every architectural decision we made.
Everything is open-source under GPL-v3: model weights, pre-training code, the full 350-million-cell dataset (aligned into an atlas with 1% available for interactive exploration), pre-training task implementations, and the weights for all 42 models in the ablation study. We open-sourced the models we didn’t use, not just the winner.
scPRINT-2 is also built for practical use. Inference on a dataset of 100,000 cells runs in minutes on a single GPU. The model handles variable gene context lengths — from 200 to 8,000 genes — without retraining, because it was trained with variable context from the start.
The paper is available on bioRxiv. The code and weights are on GitHub. In the companion posts, we go deeper on the additive study and what it taught us about scFM design, the assembly of our 350-million-cell corpus, and the Criss-Cross Attention mechanism we developed to break the quadratic bottleneck in gene-level attention.
scPRINT-2 is available at github.com/cantinilab/scPRINT-2 under GPL-v3. Read the companion posts: 350 Million Cells, 16 Organisms · Criss-Cross Attention.



Leave a comment