
The Gene Regulation Landscape
I have always wanted to understand how a cell works.
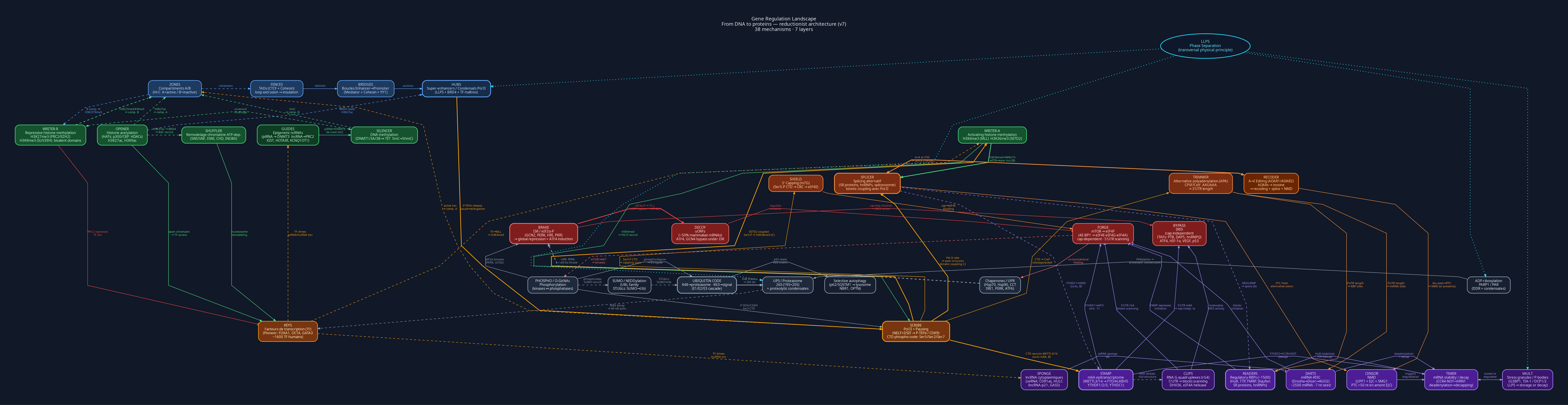
Most of the time, even when textbooks go into molecular detail, the story is still organized around dogmas. One chapter adds chromatin. Another adds transcription factors. Later, RNA processing appears. Then translation, protein degradation, signaling, condensates, non-coding RNAs, and so on. Each new mechanism is real, but they often arrive as separate layers of complexity, not as one formal picture of what we collectively know.
That is what I tried to build here: a summary of our current formal knowledge of gene regulation, placed into one landscape.
While doing it, I noticed something that surprised me. Biology has many measurements and many local names, but sometimes no clean conceptual object for things that are probably views of the same underlying cellular structure. For example, super-enhancers in ChIP-seq and transcriptional condensates in microscopy are not strictly identical, but they are clearly not unrelated either. In other places, molecular biology has long descriptive sentences for mechanisms, but no short handle that makes the mechanism easy to reason about.
So I gave each main mechanism a short code name. The names are not meant to replace the biology; they are handles that point back to precise glossary entries. I hope the map is useful, and maybe sparks discussions.
The map is very large, so the embedded version below is mostly a preview. You can open the full-resolution zoomable map here. The companion technical notes contain the 1-to-1 glossary for every box name, the full mechanism catalogue, link rationale, and legend details. The Graphviz DOT source is also available. A node-by-node literature audit now reviews the biological support, caveats, and proposed graph revisions for all 37 mechanism boxes. Several arrows were added or softened after that audit: for example, the cap now links to mRNA stability, growth and translation state now regulate decoding tempo, codon optimality and translation surveillance now link to mRNA decay, chaperone triage links back to ubiquitin routing, signaling links to CRL licensing, and nuclear lncRNA guides link to repressive histone writers. Other arrows are deliberately dashed because the evidence is contextual rather than universal.
In the figure, each box title is a code name that maps 1-to-1 to the catalogue
entry in the technical notes. Solid arrows are the main mechanistic relations;
dashed arrows indicate contextual, feedback, or association-style links;
tee-headed arrows indicate repression; bold arrows mark especially important
coupling edges. Border styles mark meta-principles such as LLPS and DECAY:
these are recurring physical or regulatory motifs that appear across several
mechanisms, not separate boxes in the pathway.
The map is also intentionally reductionist. I did not try to draw every important named event as its own box. Some biological phenomena are real and important, but they are better understood as outputs of several lower-level mechanisms already shown in the graph. For example, promoter/TSS choice depends on transcription-factor grammar, chromatin accessibility, enhancer contacts, and Pol II initiation/pausing. Transcription termination depends on Pol II state, cleavage/polyadenylation, chromatin context, and RNA decay machinery. Whether an RNA leaves the nucleus depends on capping, splicing, 3’ processing, RNA-binding proteins, RNA marks, and quality control. In those cases, the goal is not to add a new named event whenever biology has a label for one. The goal is to ask whether the underlying decision mechanisms and their links are already represented.
This is also why some familiar topics, such as R-loops, DNA torsional stress, transcription-replication conflicts, nuclear bodies, broad epitranscriptomic marks beyond m6A, repeats/transposons, motif grammar, and Pol I/Pol III rRNA/tRNA biology are handled as caveats, extensions, or submechanisms in the technical notes rather than as new boxes in the main landscape. That choice is not a claim that they are unimportant. It is a claim about map resolution: this figure prioritizes reusable regulatory mechanisms over named composite events.
Post-translational control is shown with representative feedback routes rather
than every substrate-specific event. SWITCH, ROUTER, and DESTROY can touch
many upstream programs; the map draws the main recurring routes to TF activity,
Pol II, stress translation, proteostasis, and mTOR/NF-κB-style feedback.
LICENSE is intentionally narrower: NEDDylation mainly activates cullin-RING E3
ligases, so its main graph role is to feed ROUTER.
Box Glossary
Each box in the image has exactly one entry here. Box titles are the short code names; italic text inside the image marks genes or proteins when the label needs examples.
| Code | Layer | Meaning |
|---|---|---|
ZONES |
3D genome | A/B chromatin compartments. |
FENCES |
3D genome | TAD boundaries and insulation by CTCF/cohesin. |
BRIDGES |
3D genome | Enhancer-promoter loops. |
HUBS |
3D genome | Super-enhancers / enhancer hubs, with Pol II/coactivator condensates treated as a related but not identical physical model. |
SILENCER |
Epigenetics | DNA methylation and repressive chromatin memory. |
OPENER |
Epigenetics | Histone acetylation that opens chromatin. |
WRITER-A |
Epigenetics | Activating histone methylation marks. |
WRITER-R |
Epigenetics | Repressive histone methylation marks. |
SHUFFLER |
Epigenetics | ATP-dependent nucleosome remodeling. |
GUIDES |
Epigenetics | ncRNAs that recruit chromatin regulators. |
KEYS |
Transcription | Transcription factors, including pioneer factors. |
SCRIBE |
Transcription | Pol II pausing, release, and CTD phosphorylation. |
SHIELD |
Co-transcriptional | 5’ capping and cap-dependent protection/export. |
SPLICER |
Co-transcriptional | Alternative splicing coupled to Pol II kinetics. |
TRIMMER |
Co-transcriptional | Alternative polyadenylation and 3’UTR choice. |
RECODER |
Co-transcriptional | A-to-I RNA editing by ADARs. |
STAMP |
Post-transcriptional | m6A RNA marking and reader-dependent fate choices. |
READERS |
Post-transcriptional | RNA-binding proteins that tune RNA processing, stability, localization, and translation. |
DARTS |
Post-transcriptional | miRNA-RISC targeting. |
SPONGE |
Post-transcriptional | Cytoplasmic lncRNA/circRNA competition with miRNAs, kept as a strongly stoichiometry- and localization-dependent mechanism. |
CENSOR |
Post-transcriptional | Nonsense-mediated mRNA decay. |
TIMER |
Post-transcriptional | mRNA half-life, deadenylation, decapping, and decay. |
CLIPS |
Post-transcriptional | RNA G-quadruplex structures that affect scanning and translation. |
VAULT |
Post-transcriptional | Stress granules and P-bodies for RNA storage or decay. |
FORGE |
Translation | Starting cap-dependent translation through mTOR/eIF4F. |
BRAKE |
Translation | Slowing global translation during stress through ISR/eIF2α-P. |
DECOY |
Translation | uORFs that divert scanning ribosomes and gate main ORF translation. |
BYPASS |
Translation | Non-canonical initiation routes; viral IRESs are robust, while many cellular IRES-like claims need strict controls. |
TEMPO |
Translation | Decoding kinetics, tRNA/codon effects, ribosome state, and their regulation by growth, initiation load, and stress context. Also covers decoding fidelity: alternate decoding can install non-genomic amino-acid substitutions that yield stable, abundant proteoforms. |
INSPECTOR |
Translation | RQC/NGD/NSD surveillance of stalled or broken translation. |
SWITCH |
Post-translational | Phosphorylation/O-GlcNAc switches for protein activity and interactions. |
ROUTER |
Post-translational | Ubiquitin-chain logic that routes proteins to signaling, proteasome, or autophagy. |
TETHER |
Post-translational | SUMOylation that tethers proteins into nuclear complexes, repression modules, or repair assemblies. |
LICENSE |
Post-translational | Neddylation that licenses cullin-RING E3 ubiquitin ligases, regulated by CRL assembly and signaling context. |
DESTROY |
Post-translational | Protein clearance through two fused outputs: proteasome and selective autophagy. |
MATURE |
Post-translational | Protein folding, refolding, triage, and ER-stress UPR. |
PAR |
Post-translational | PARP/PAR signaling at DNA damage and repair condensates. |
Term Glossary
These are the main non-gene, non-protein terms used in the figure and glossary.
| Term | Meaning |
|---|---|
| A/B compartments | Large Hi-C chromatin domains; A is generally active/euchromatic, B is generally inactive/heterochromatic. |
| TAD | Topologically associating domain; a chromatin neighborhood insulated by boundaries such as CTCF/cohesin sites. |
| Enhancer-promoter loop | A 3D contact that brings a distal enhancer near a target promoter. |
| Super-enhancer | A dense enhancer cluster with high transcription-factor, Mediator, BRD4, and Pol II occupancy; an operational enhancer annotation, not automatically proof of a condensate. |
| Condensate / LLPS | Liquid-liquid phase separation; concentration of molecules into a dense phase without a membrane. |
| DNA methylation | Addition of methyl groups to cytosines, often linked to transcriptional repression at promoters. |
| Histone mark | A chemical modification on histones, such as acetylation or methylation, read by chromatin proteins. |
| Chromatin remodeling | ATP-driven repositioning, eviction, or exchange of nucleosomes. |
| ncRNA | Non-coding RNA; RNA that functions without being translated into protein. |
| Pol II pausing | Promoter-proximal RNA polymerase II stalling before productive elongation. |
| CTD phosphorylation | Phosphorylation of the Pol II C-terminal domain, coordinating transcription with RNA processing. |
| 5’ capping | Addition of an m7G cap to nascent RNA, protecting it and enabling export/translation. |
| Alternative splicing | Regulated exon choice that produces multiple transcript isoforms from one gene. |
| Alternative polyadenylation | Choice of different cleavage/polyA sites, often changing 3’UTR length. |
| A-to-I editing | Adenosine-to-inosine RNA editing; inosine is read like guanosine by many machines. |
| m6A | N6-methyladenosine, a reversible RNA modification interpreted by reader proteins. |
| RBP | RNA-binding protein. |
| miRNA-RISC | MicroRNA loaded into the RISC complex to repress or destabilize target RNAs. |
| ceRNA | Competing endogenous RNA; an RNA that can buffer miRNAs by sharing target sites, when abundance, affinity, and colocalization are sufficient. |
| NMD | Nonsense-mediated decay; surveillance and degradation of transcripts with premature stop codons. |
| Deadenylation / decapping | Removal of the polyA tail and 5’ cap, usually committing an mRNA to decay. |
| RNA G-quadruplex | A guanine-rich RNA structure that can block scanning or alter RNA fate. |
| Stress granule / P-body | Cytoplasmic RNA-protein condensates involved in RNA storage, repression, or decay. |
| Cap-dependent translation | Canonical translation initiation through cap recognition and 5’UTR scanning. |
| ISR / eIF2α-P | Integrated stress response; phosphorylation of eIF2α lowers global initiation but favors selected mRNAs. |
| uORF | Upstream open reading frame in a 5’UTR that can divert scanning ribosomes. |
| IRES / ITAF | Internal ribosome entry site and its helper factors. Viral IRESs are strong examples; many cellular IRES claims need strict controls for cryptic promoters, splicing, readthrough, and RNA abundance. |
| RQC / NGD / NSD | Ribosome quality control, no-go decay, and non-stop decay; surveillance of stalled or abnormal translation. |
| O-GlcNAc | Reversible sugar modification on Ser/Thr residues that can crosstalk with phosphorylation. |
| Ubiquitin chain | A polymeric ubiquitin mark whose linkage type, such as K48 or K63, helps determine protein fate. |
| SUMOylation | Conjugation of SUMO proteins, often changing nuclear interactions or complex assembly. |
| Neddylation | Conjugation of NEDD8, especially to cullins, activating cullin-RING E3 ligases. |
| Proteasome | Protease complex that degrades many short-lived or damaged ubiquitinated proteins. |
| Selective autophagy | Lysosomal clearance of selected cargo such as aggregates, organelles, or ubiquitinated complexes. |
| UPR | Unfolded protein response; ER-stress response that expands folding capacity or slows translation. |
| PAR / ADP-ribosylation | Poly-ADP-ribose signaling, often used around DNA damage and condensate formation. |
The Seven Layers
The map follows the flow from DNA to RNA to protein:
- 3D genome:
ZONES,FENCES,BRIDGES, andHUBSrepresent A/B compartments, TADs, enhancer-promoter loops, and super-enhancers that can overlap with transcriptional condensates without being identical to them. - Epigenetics:
SILENCER,OPENER,WRITER-A,WRITER-R,SHUFFLER, andGUIDEScover DNA methylation, histone marks, chromatin remodeling, and non-coding RNAs that guide chromatin complexes. - Transcription:
KEYSare transcription factors;SCRIBEis Pol II, promoter-proximal pausing, and the phosphorylation code of its CTD. - Co-transcriptional processing:
SHIELD,SPLICER,TRIMMER, andRECODERcover capping, alternative splicing, alternative polyadenylation, and A-to-I RNA editing. - Post-transcriptional control:
STAMP,READERS,DARTS,SPONGE,CENSOR,TIMER,CLIPS, andVAULTcover m6A, RNA-binding proteins, miRNAs, lncRNAs, nonsense-mediated decay, mRNA stability, RNA structures, and cytoplasmic granules. - Translation:
FORGE,BRAKE,DECOY,BYPASS,TEMPO, andINSPECTORdescribe cap-dependent initiation, the integrated stress response, uORFs, non-canonical initiation, decoding kinetics, and ribosome quality control.TEMPOnow also folds in decoding fidelity: recent proteogenomics across >1,000 human samples found thousands of non-genomic amino-acid substitutions from alternate ribosomal decoding — not explained by DNA variants or A-to-I editing — producing proteoforms that are stable, abundant, and tissue/cancer-specific (Tsour et al., Nature 2026). Because these proteins partly escapeINSPECTORsurveillance and persist intoMATURE, this belongs on the speed-vs-fidelity axis ofTEMPOrather than inRECODER(which is transcript-level A-to-I editing). - Post-translational regulation:
SWITCH,ROUTER,TETHER,LICENSE,DESTROY,MATURE, andPARcover phosphorylation/O-GlcNAc, ubiquitin, SUMOylation, neddylation, proteasome/autophagy clearance, maturation/UPR, and PARP/PAR signaling.
Across the whole diagram, border styles flag recurring meta-principles. LLPS
marks liquid-liquid phase separation, and DECAY marks turnover or clearance.
They are not extra regulatory layers. They are reused in several places:
transcriptional condensates, stress granules and P-bodies, mRNA decay,
proteolytic condensates, autophagy, and DNA damage repair assemblies.
The Useful Reduction
The full map contains 37 mechanism boxes, 2 meta-principles, and dozens of interactions. But conceptually, most of gene regulation reduces to three strategies.
1. Control accessibility.
Make a substrate accessible or inaccessible to its molecular machinery.
Chromatin opening lets transcription factors bind. TADs constrain which
enhancers can contact which promoters. Stress granules temporarily remove mRNAs
from translation. miRNAs and lncRNAs tune whether an mRNA is available to the
ribosome.
2. Write a reversible mark, then interpret it.
Histone methylation, DNA methylation, m6A, phosphorylation, ubiquitination,
SUMOylation: the mark alone is never the full story. The reader and the context
determine the output. m6A can promote translation or accelerate decay. A K48
ubiquitin chain points toward the proteasome; K63 often acts in signaling or
selective autophagy. Phosphorylation can activate a transcription factor or
create a degron.
3. Couple two processes through kinetics.
Some regulation is not a static state but a timing problem. Pol II elongation
speed influences exon choice. SETD2 deposits H3K36me3 during elongation, linking
transcription to splicing. eIF2α phosphorylation globally slows translation but
selectively favors ATF4 through uORF logic. Codon usage changes ribosome speed
and can influence co-translational folding.
That is the central idea of the landscape: gene regulation is not just a list of mechanisms. It is a multi-layer control architecture built from recurring design patterns.
Why This Matters For AI Biology
For AI biology, this kind of map is not only educational. It shows why predicting “gene expression” cannot be reduced to reading a promoter sequence.
The output of a gene depends on chromatin state, 3D contacts, Pol II kinetics, splicing, RNA modifications, RNA-binding proteins, translational control, and protein lifetime. A model that wants to predict perturbation response, cell state, or disease mechanism needs to represent at least part of this stack.
The lesson of the Gene Regulation Landscape is simple: gene expression is not a scalar. It is the endpoint of a control system.
Sources To Anchor The Map
- Core & Adelman, 2019, promoter-proximal Pol II pausing: https://pubmed.ncbi.nlm.nih.gov/31123063/
- Naftelberg et al., 2015, transcription/chromatin/splicing coupling: https://pubmed.ncbi.nlm.nih.gov/26034889/
- Wang & He, 2014, dynamic RNA modifications: https://pubmed.ncbi.nlm.nih.gov/25263552/
- Wang et al., 2015, m6A and translation efficiency: https://www.cell.com/cell/fulltext/S0092-8674(15)00562-0
- Shi et al., 2017, YTHDF3 translation/decay: https://pmc.ncbi.nlm.nih.gov/articles/PMC5339834/
- Sabari et al., 2018, coactivator condensation at super-enhancers: https://pmc.ncbi.nlm.nih.gov/articles/PMC6092193/
- Robson et al., 2019, chromatin topology: https://pubmed.ncbi.nlm.nih.gov/31324893/



Leave a comment