PyCUB
During my master thesis I had the chance to work with It a multidisciplinary team involving teachers in CS and Biology, 🧪 and a PhD student. I had the chance to work on sets of data from EMBL and from our lab at the University of Kent.
 Artist’s view of our genes.
Artist’s view of our genes.
It is a 6 month long project involving a lot of data science, computer, 💻 science and comparative genomics, 🧬 . I worked mostly on fungi and tried many different paths.
I found back many of the results from the literature regarding the CUB. I put into question on of the recent results published in Nature. I build an entire software and toolkit for the analysis of the Codon Usage Bias and proved its relevance with the aforementioned results.
You can find more information about this project and how to use it on the Github repository.
The documentation is also available Here.
.](https://cdn-images-1.medium.com/max/4000/1*-Ih-Pp12oQIsjw7PKSHbLQ.png) have a look at the full Master Thesis.
have a look at the full Master Thesis.
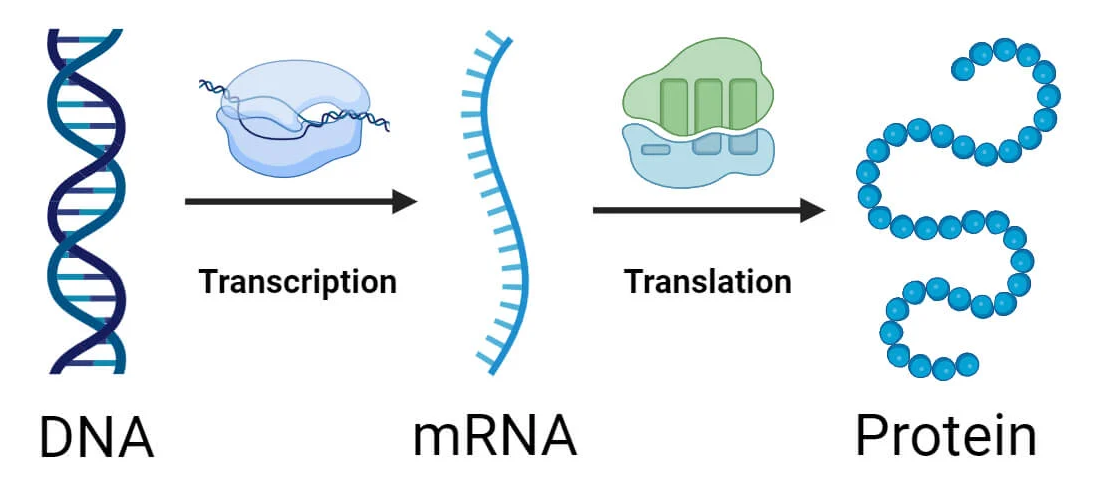
The codon usage bias is the DNA sequence pattern problem in protein coding genes, where sets of synonymous codons, blocks of 3 nucleotides which encode for the same amino acid in the transcription process, have a non random distribution.
As codons are very low level features in the genome, many factors might explain their particular distributions. Amongst the stated ones, the GC content of the genome, the tRNA pool, the replication speed of the cell, as well as the size, the evolutionary distance and pressure over the gene might all have a degree of influence over it.
How much does each factor help to explain the bias registered in the codon usage of genes? How much of the codon usage bias are they able to predict? We set out to study such questions using advanced statistical tools, over a set of related species.
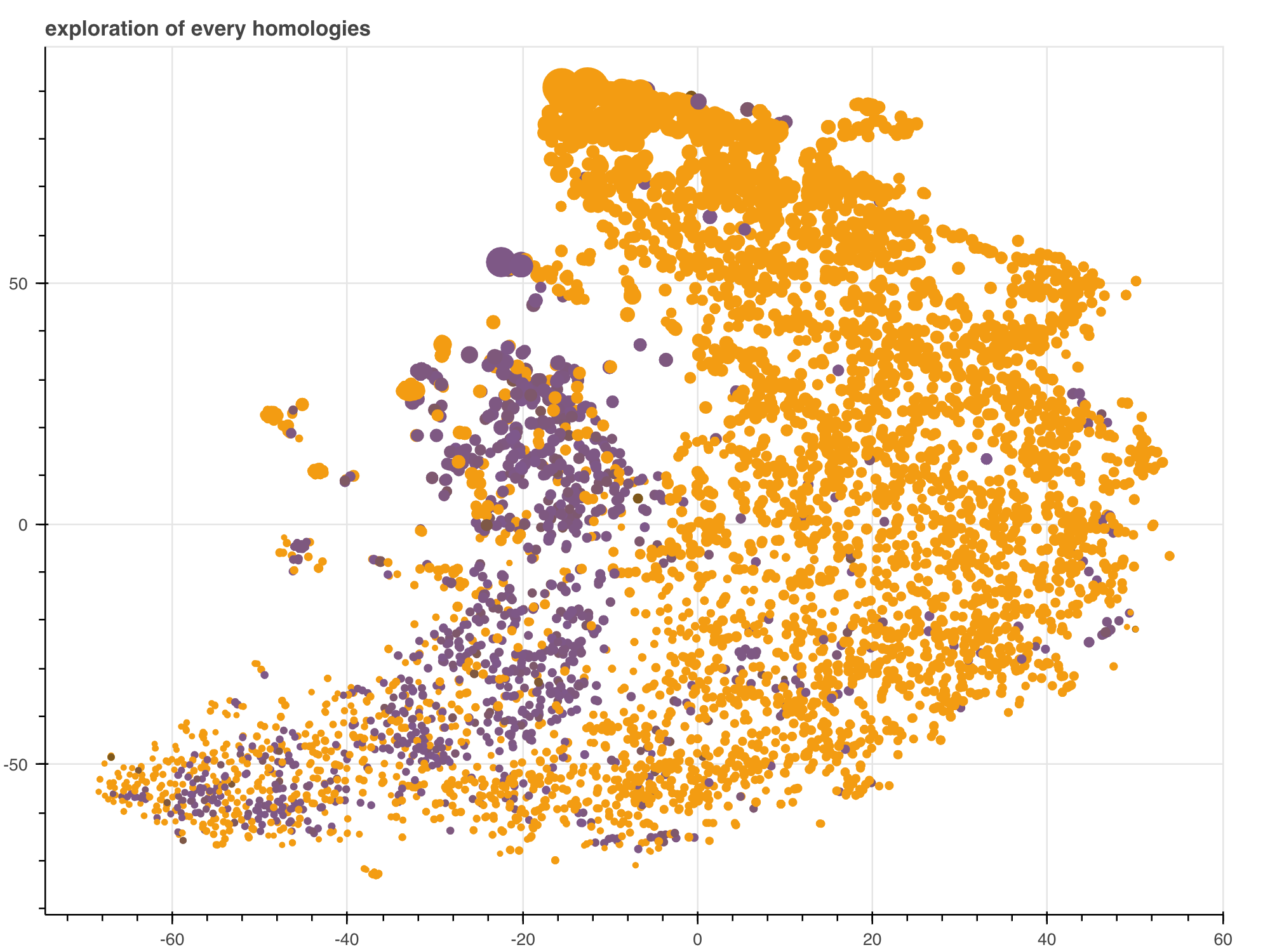
 A heatmap plot of the entropy cost of each coding sequences for each species with tSNE.
A heatmap plot of the entropy cost of each coding sequences for each species with tSNE.



Leave a comment